A journal of IEEE and CAA , publishes
high-quality papers in English on original
theoretical/experimental research
and development in all areas of automation
 Volume 11
Issue 10
Volume 11
Issue 10
IEEE/CAA Journal of Automatica Sinica
| Citation: | G. Li, B. Zhao, X. Su, D. Li, Y. Yang, Z. Zeng, and L. Hu, “Learning sequential and structural dependencies between nucleotides for RNA N6-methyladenosine site identification,” IEEE/CAA J. Autom. Sinica, vol. 11, no. 10, pp. 2123–2134, Oct. 2024. doi: 10.1109/JAS.2024.124233

|
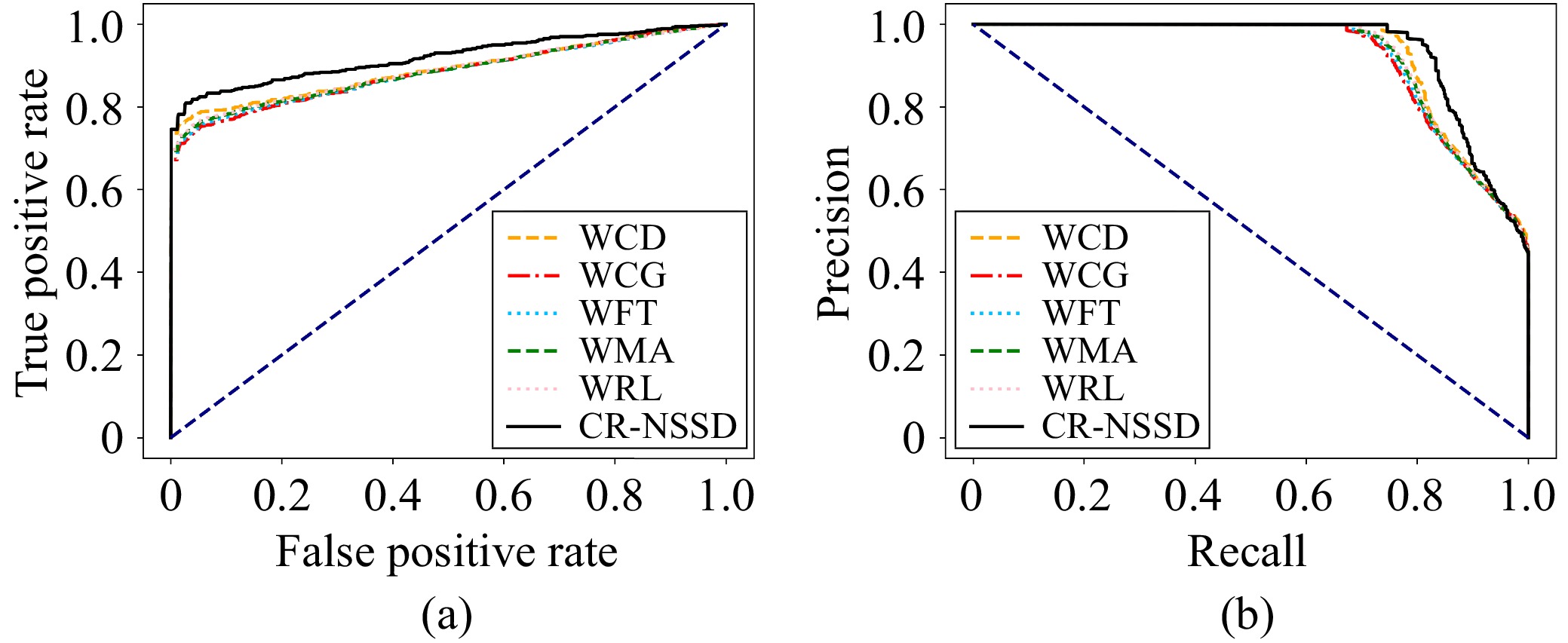
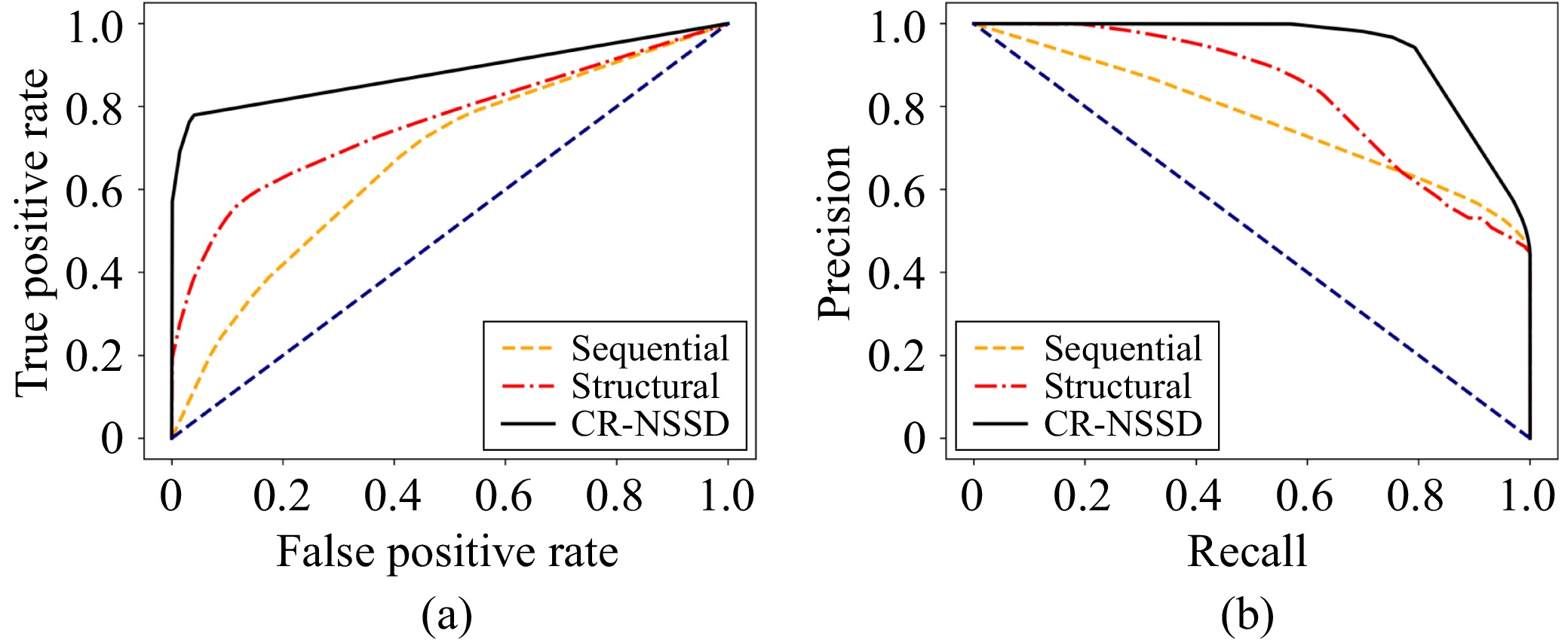
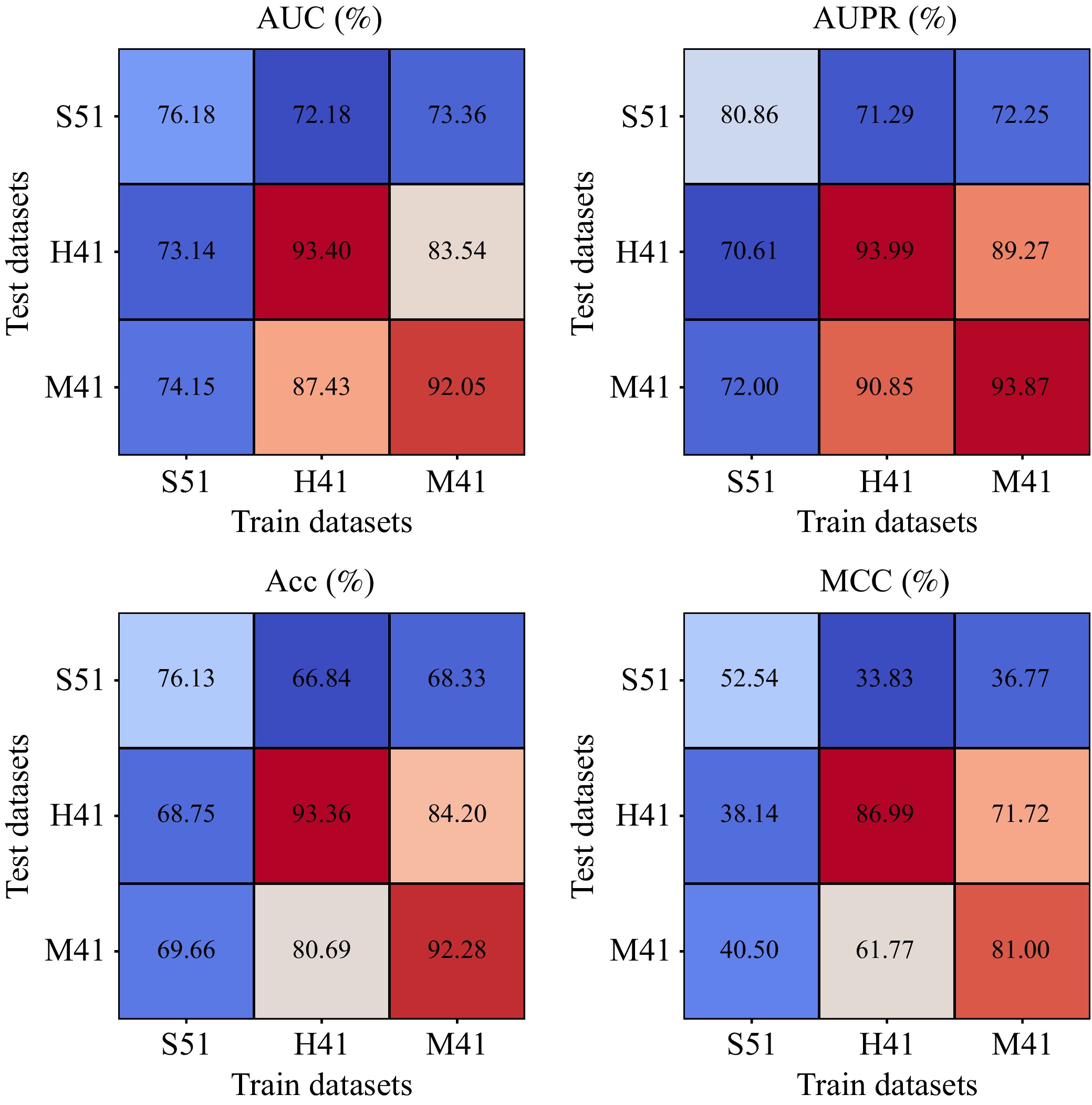
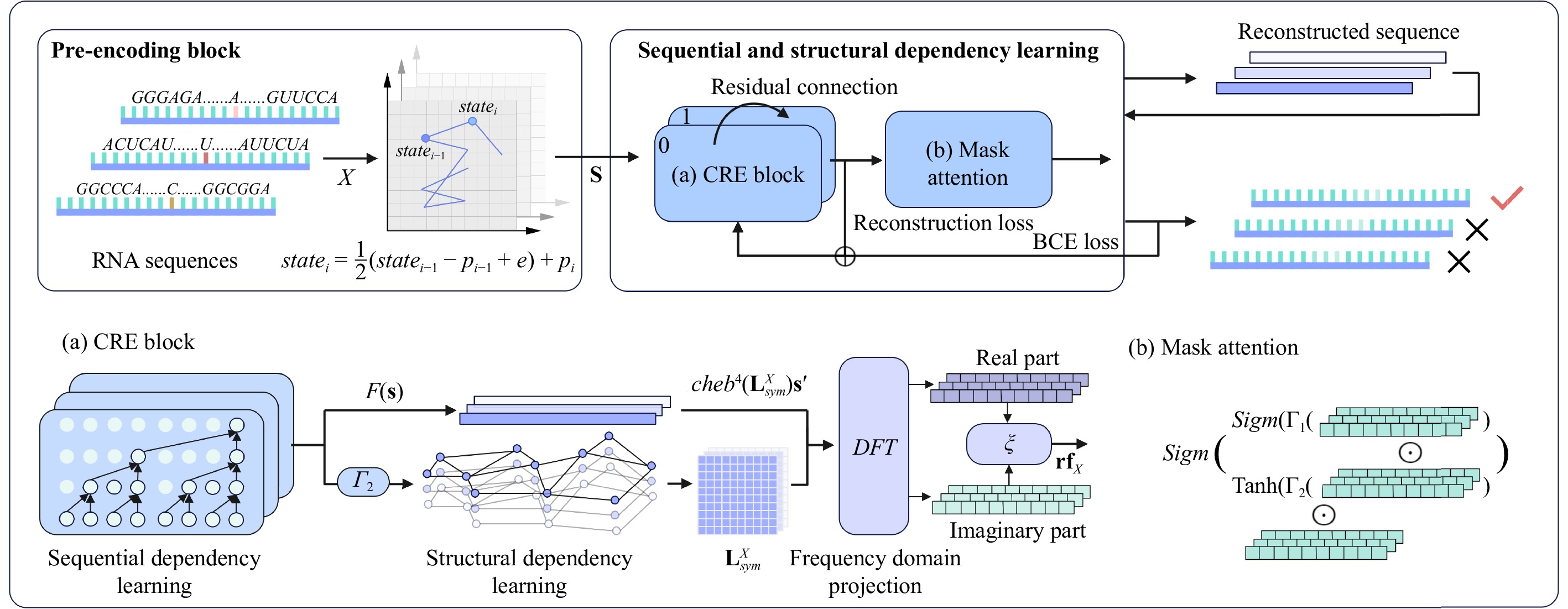
N6-methyladenosine (m6A) is an important RNA methylation modification involved in regulating diverse biological processes across multiple species. Hence, the identification of m6A modification sites provides valuable insight into the biological mechanisms of complex diseases at the post-transcriptional level. Although a variety of identification algorithms have been proposed recently, most of them capture the features of m6A modification sites by focusing on the sequential dependencies of nucleotides at different positions in RNA sequences, while ignoring the structural dependencies of nucleotides in their three-dimensional structures. To overcome this issue, we propose a cross-species end-to-end deep learning model, namely CR-NSSD, which conduct a cross-domain representation learning process integrating nucleotide structural and sequential dependencies for RNA m6A site identification. Specifically, CR-NSSD first obtains the pre-coded representations of RNA sequences by incorporating the position information into single-nucleotide states with chaos game representation theory. It then constructs a cross-domain reconstruction encoder to learn the sequential and structural dependencies between nucleotides. By minimizing the reconstruction and binary cross-entropy losses, CR-NSSD is trained to complete the task of m6A site identification. Extensive experiments have demonstrated the promising performance of CR-NSSD by comparing it with several state-of-the-art m6A identification algorithms. Moreover, the results of cross-species prediction indicate that the integration of sequential and structural dependencies allows CR-NSSD to capture general features of m6A modification sites among different species, thus improving the accuracy of cross-species identification.
| [1] |
H.-C. Duan, Y. Wang, and G. Jia, “Dynamic and reversible RNA N6-methyladenosine methylation,” Wiley Interdiscip Rev RNA, vol. 10, no. 1, p. e1507, 2019. doi: 10.1002/wrna.1507
|
| [2] |
Z. Xu, B. Lv, Y. Qin, and B. Zhang, “Emerging roles and mechanism of m6a methylation in cardiometabolic diseases,” Cells, vol. 11, no. 7, p. 1101, 2022. doi: 10.3390/cells11071101
|
| [3] |
K. D. Meyer and S. R. Jaffrey, “The dynamic epitranscriptome: N6-methyladenosine and GENE expression control,” Nat. Rev. Mol. Cell Biol., vol. 15, no. 5, pp. 313–326, 2014.
|
| [4] |
S. Geula, S. Moshitch-Moshkovitz, D. Dominissini, et al., “m6A mRNA methylation facilitates resolution of naïve pluripotency toward differentiation,” Science, vol. 347, no. 6225, pp. 1002–1006, 2015. doi: 10.1126/science.1261417
|
| [5] |
S. A. Hinger, J. Wei, L. E. Dorn, B. A. Whitson, P. M. Janssen, C. He, and F. Accornero, “Remodeling of the m6a landscape in the heart reveals few conserved post-transcriptional events underlying cardiomyocyte hypertrophy,” J. Mol. Cell. Cardiol., vol. 151, pp. 46–55, 2021. doi: 10.1016/j.yjmcc.2020.11.002
|
| [6] |
Y. Qin, L. Li, E. Luo, J. Hou, G. Yan, D. Wang, Y. Qiao, and C. Tang, “Role of m6A RNA methylation in cardiovascular disease,” Int. J. Mol. Med., vol. 46, no. 6, pp. 1958–1972, 2020. doi: 10.3892/ijmm.2020.4746
|
| [7] |
J. Liang, J. Sun, W. Zhang, X. Wang, Y. Xu, Y. Peng, L. Zhang, W. Xiong, Y. Liu, and H. Liu, “Novel insights into the roles of N6-methyladenosine (m6A) modification and autophagy in human diseases,” Int. J. Biol. Sci., vol. 19, no. 2, p. 705, 2023. doi: 10.7150/ijbs.75466
|
| [8] |
K. D. Meyer, Y. Saletore, P. Zumbo, O. Elemento, C. E. Mason, and S. R. Jaffrey, “Comprehensive analysis of mRNA methylation reveals enrichment in 3’utrs and near stop codons,” Cell, vol. 149, no. 7, pp. 1635–1646, 2012. doi: 10.1016/j.cell.2012.05.003
|
| [9] |
D. Dominissini, S. Moshitch-Moshkovitz, S. Schwartz, et al., “Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq,” Nature, vol. 485, no. 7397, pp. 201–206, 2012. doi: 10.1038/nature11112
|
| [10] |
W. Chen, H. Tran, Z. Liang, H. Lin, and L. Zhang, “Identification and analysis of the N6-methyladenosine in the saccharomyces cerevisiae transcriptome,” Sci. Rep., vol. 5, no. 1, pp. 1–8, 2015.
|
| [11] |
Z. Song, D. Huang, B. Song, K. Chen, Y. Song, G. Liu, J. Su, J. P. d. Magalhães, D. J. Rigden, and J. Meng, “Attention-based multi-label neural networks for integrated prediction and interpretation of twelve widely occurring RNA modifications,” Nat. Commun., vol. 12, no. 1, pp. 1–11, 2021. doi: 10.1038/s41467-020-20314-w
|
| [12] |
Y. Zhang and M. Hamada, “DeepM6ASeq: Prediction and characterization of m6A-containing sequences using deep learning,” BMC Bioinform., vol. 19, no. 19, pp. 1–11, 2018.
|
| [13] |
W.-R. Qiu, S.-Y. Jiang, B.-Q. Sun, X. Xiao, X. Cheng, and K.-C. Chou, “IRNA-2Methyl: Identify RNA 2’-o-methylation sites by incorporating sequence-coupled effects into general PSEKNC and ensemble classifier,” Med. Chem., vol. 13, no. 8, pp. 734–743, 2017.
|
| [14] |
H. Yang, H. Lv, H. Ding, W. Chen, and H. Lin, “iRNA-2OM: A sequence-based predictor for identifying 2’-o-methylation sites in Homo sapiens,” J. Comput. Biol., vol. 25, no. 11, pp. 1266–1277, 2018. doi: 10.1089/cmb.2018.0004
|
| [15] |
W. Chen, H. Ding, X. Zhou, H. Lin, and K.-C. Chou, “iRNA (m6A)-PSEDNC: Identifying n6-methyladenosine sites using pseudo dinucleotide composition,” Anal. Biochem., vol. 561, pp. 59–65, 2018.
|
| [16] |
W. Chen, P. Feng, H. Ding, H. Lin, and K.-C. Chou, “IRNA-Methyl: Identifying N6-methyladenosine sites using pseudo nucleotide composition,” Anal. Biochem., vol. 490, pp. 26–33, 2015. doi: 10.1016/j.ab.2015.08.021
|
| [17] |
W.-R. Qiu, S.-Y. Jiang, Z.-C. Xu, X. Xiao, and K.-C. Chou, “iRNAm5C-PseDNC: Identifying RNA 5-methylcytosine sites by incorporating physical-chemical properties into pseudo dinucleotide composition,” Oncotarget, vol. 8, no. 25, p. 41178, 2017. doi: 10.18632/oncotarget.17104
|
| [18] |
W. Chen, X. Song, H. Lv, and H. Lin, “iRNA-m2G: Identifying N2-methylguanosine sites based on sequence-derived information,” Mol. Ther. Nucleic. Acids, vol. 18, pp. 253–258, 2019. doi: 10.1016/j.omtn.2019.08.023
|
| [19] |
W. Chen, P. Feng, X. Song, H. Lv, and H. Lin, “iRNA-m7G: Identifying N7-methylguanosine sites by fusing multiple features,” Mol. Ther. Nucleic. Acids, vol. 18, pp. 269–274, 2019. doi: 10.1016/j.omtn.2019.08.022
|
| [20] |
M. Tahir, H. Tayara, and K. T. Chong, “iRNA-PseKNC (2Methyl): Identify RNA 2’-o-methylation sites by convolution neural network and chou’s pseudo components,” J. Theor. Biol., vol. 465, pp. 1–6, 2019. doi: 10.1016/j.jtbi.2018.12.034
|
| [21] |
W. Chen, H. Tang, J. Ye, H. Lin, and K.-C. Chou, “IRNA-PSEU: Identifying RNA pseudouridine sites,” Mol. Ther. Nucleic. Acids, vol. 5, p. e332, 2016.
|
| [22] |
D. M. Karl, “Cellular nucleotide measurements and applications in microbial ecology,” Microbiological Reviews, vol. 44, no. 4, pp. 739–796, 1980. doi: 10.1128/mr.44.4.739-796.1980
|
| [23] |
Y. Zhou, P. Zeng, Y.-H. Li, Z. Zhang, and Q. Cui, “Sramp: Prediction of mammalian N6-methyladenosine (m6a) sites based on sequencederived features,” Nucleic Acids Res., vol.44, no.10, p. e91, 2016.
|
| [24] |
X. Qiang, H. Chen, X. Ye, R. Su, and L. Wei, “M6AMRFS: Robust prediction of N6-methyladenosine sites with sequence-based features in multiple species,” Front. Genet., vol. 9, p. 495, 2018. doi: 10.3389/fgene.2018.00495
|
| [25] |
L. E. Peterson, “K-nearest neighbor,” Scholarpedia, vol. 4, no. 2, p. 1883, 2009. doi: 10.4249/scholarpedia.1883
|
| [26] |
Z. Chen, P. Zhao, F. Li, Y. Wang, A. I. Smith, G. I. Webb, T. Akutsu, A. Baggag, H. Bensmail, and J. Song, “Comprehensive review and assessment of computational methods for predicting RNA post-transcriptional modification sites from RNA sequences,” Brief. Bioinform., vol. 21, no. 5, pp. 1676–1696, 2020. doi: 10.1093/bib/bbz112
|
| [27] |
Q. Zou, P. Xing, L. Wei, and B. Liu, “Gene2vec: Gene subsequence embedding for prediction of mammalian n6-methyladenosine sites from mRNA,” RNA, vol. 25, no. 2, pp. 205–218, 2019. doi: 10.1261/rna.069112.118
|
| [28] |
I. Nazari, M. Tahir, H. Tayara, and K. T. Chong, “IN6-Methyl (5-step): Identifying RNA N6-methyladenosine sites using deep learning mode via Chou’s 5-step rules and Chou’s general PSEKNC,” Chemom. Intell. Lab. Syst., vol. 193, p. 103811, 2019. doi: 10.1016/j.chemolab.2019.103811
|
| [29] |
C. Pian, Z. Yang, Y. Yang, L. Zhang, and Y. Chen, “Identifying RNA N6-methyladenine sites in three species based on a Markov model,” Front. Genet., vol. 12, p. 650803, 2021. doi: 10.3389/fgene.2021.650803
|
| [30] |
Y. Huang, N. He, Y. Chen, Z. Chen, and L. Li, “BERMP: A cross-species classifier for predicting m6a sites by integrating a deep learning algorithm and a random forest approach,” Int. J. Biol. Sci., vol. 14, no. 12, p. 1669, 2018. doi: 10.7150/ijbs.27819
|
| [31] |
F.-Y. Dao, H. Lv, Y.-H. Yang, H. Zulfiqar, H. Gao, and H. Lin, “Computational identification of N6-methyladenosine sites in multiple tissues of mammals,” Computational and Structural Biotechnology J., vol. 18, pp. 1084–1091, 2020. doi: 10.1016/j.csbj.2020.04.015
|
| [32] |
M. J. Pietal, J. M. Bujnicki, and L. P. Kozlowski, “GDFuzz3D: A method for protein 3D structure reconstruction from contact maps, based on a non-euclidean distance function,” Bioinformatics, vol. 31, no. 21, pp. 3499–3505, 2015. doi: 10.1093/bioinformatics/btv390
|
| [33] |
K. Rother, M. Rother, M. Boniecki, T. Puton, and J. M. Bujnicki, “RNA and protein 3D structure modeling: Similarities and differences,” J. Mol Model, vol. 17, no. 9, pp. 2325–2336, 2011. doi: 10.1007/s00894-010-0951-x
|
| [34] |
M. Parisien and F. Major, “The MC-fold and MC-SYM pipeline infers RNA structure from sequence data,” Nature, vol. 452, no. 7183, pp. 51–55, 2008. doi: 10.1038/nature06684
|
| [35] |
C. H. zu Siederdissen, S. H. Bernhart, P. F. Stadler, and I. L. Hofacker, “A folding algorithm for extended RNA secondary structures,” Bioinformatics, vol. 27, no. 13, pp. i129–i136, 2011. doi: 10.1093/bioinformatics/btr220
|
| [36] |
J. Wang, K. Mao, Y. Zhao, C. Zeng, J. Xiang, Y. Zhang, and Y. Xiao, “Optimization of RNA 3D structure prediction using evolutionary restraints of nucleotide–nucleotide interactions from direct coupling analysis,” Nucleic Acids Research, vol. 45, no. 11, pp. 6299–6309, 2017. doi: 10.1093/nar/gkx386
|
| [37] |
R. F. Alford, A. Leaver-Fay, J. R. Jeliazkov, et al., “The rosetta all-atom energy function for macromolecular modeling and design,” J. Chemical Theory and Computation, vol. 13, no. 6, pp. 3031–3048, 2017. doi: 10.1021/acs.jctc.7b00125
|
| [38] |
D. Cao, Y. Wang, J. Duan, et al., “Spectral temporal graph neural network for multivariate time-series forecasting,” in Proc. Adv. Neural Inf. Process. Syst., New Orleans, USA, 2020, vol. 33, pp. 17766–17778.
|
| [39] |
H. J. Jeffrey, “Chaos game representation of GENE structure,” Nucleic Acids Res., vol. 18, no. 8, pp. 2163–2170, 1990. doi: 10.1093/nar/18.8.2163
|
| [40] |
W. Chen, H. Tang, and H. Lin, “MethyRNA: A web server for identification of N6-methyladenosine sites,” J. Biomol. Struct. Dyn., vol. 35, no. 3, pp. 683–687, 2017. doi: 10.1080/07391102.2016.1157761
|
| [41] |
C. Musselman, H. M. Al-Hashimi, and I. Andricioaei, “IRED analysis of tar RNA reveals motional coupling, long-range correlations, and a dynamical hinge,” Biophys. J., vol. 93, no. 2, pp. 411–422, 2007. doi: 10.1529/biophysj.107.104620
|
| [42] |
P. Katsaloulis, T. Theoharis, W. Zheng, B. Hao, A. Bountis, Y. Almirantis, and A. Provata, “Long-range correlations of RNA polymerase II promoter sequences across organisms,” Physica A, vol. 366, pp. 308–322, 2006. doi: 10.1016/j.physa.2005.10.019
|
| [43] |
A. Vaswani, N. Shazeer, N. Parmar, J. Uszkoreit, L. Jones, A. N. Gomez, Ł. Kaiser, and I. Polosukhin, “Attention is all you need,” in Proc. Adv. Neural Inf. Process. Syst., Long Beach, USA, 2017, vol. 30.
|
| [44] |
K. Wang, J. Peng, and C. Yi, “The m6A consensus motif provides a paradigm of epitranscriptomic studies,” Biochemistry, vol. 60, no. 46, pp. 3410–3412, 2021. doi: 10.1021/acs.biochem.1c00254
|
| [45] |
B. Linder, A. V. Grozhik, A. O. Olarerin-George, C. Meydan, C. E. Mason, and S. R. Jaffrey, “Single-nucleotide-resolution mapping of m6A and m6Am throughout the transcriptome,” Nature Methods, vol. 12, no. 8, pp. 767–772, 2015. doi: 10.1038/nmeth.3453
|
| [46] |
C. Lea, M. D. Flynn, R. Vidal, A. Reiter, and G. D. Hager, “Temporal convolutional networks for action segmentation and detection,” in Proc. IEEE Conf. Computer Vision and Pattern Recognition, 2017, pp. 156–165.
|
| [47] |
S. Qiu, R. Liu, and Y. Liang, “GR-m6a: Prediction of N6-methyladenosine sites in mammals with molecular graph and residual network,” Computers in Biology and Medicine, vol. 163, p. 107202, 2023. doi: 10.1016/j.compbiomed.2023.107202
|
| [48] |
T. N. Kipf and M. Welling, “Semi-supervised classification with graph convolutional networks,” arXiv preprint arXiv: 1609.02907, 2016.
|
| [49] |
S. Winograd, “On computing the discrete fourier transform,” Math. Comput., vol. 32, no. 141, pp. 175–199, 1978. doi: 10.1090/S0025-5718-1978-0468306-4
|
| [50] |
M. Ilse, J. Tomczak, and M. Welling, “Attention-based deep multiple instance learning,” in Proc. Int. Conf. Mach. Learn. Stockholmsmässan, Stockholm, Sweden, P., 2018, pp. 2127–2136.
|
| [51] |
D. M. Allen, “Mean square error of prediction as a criterion for selecting variables,” Technometrics, vol. 13, no. 3, pp. 469–475, 1971. doi: 10.1080/00401706.1971.10488811
|
| [52] |
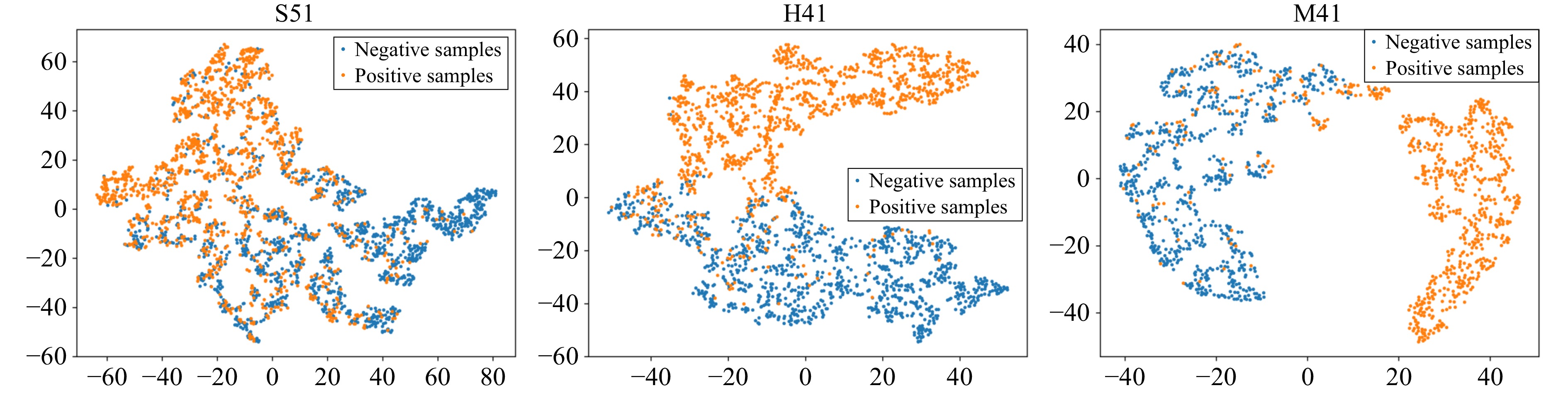
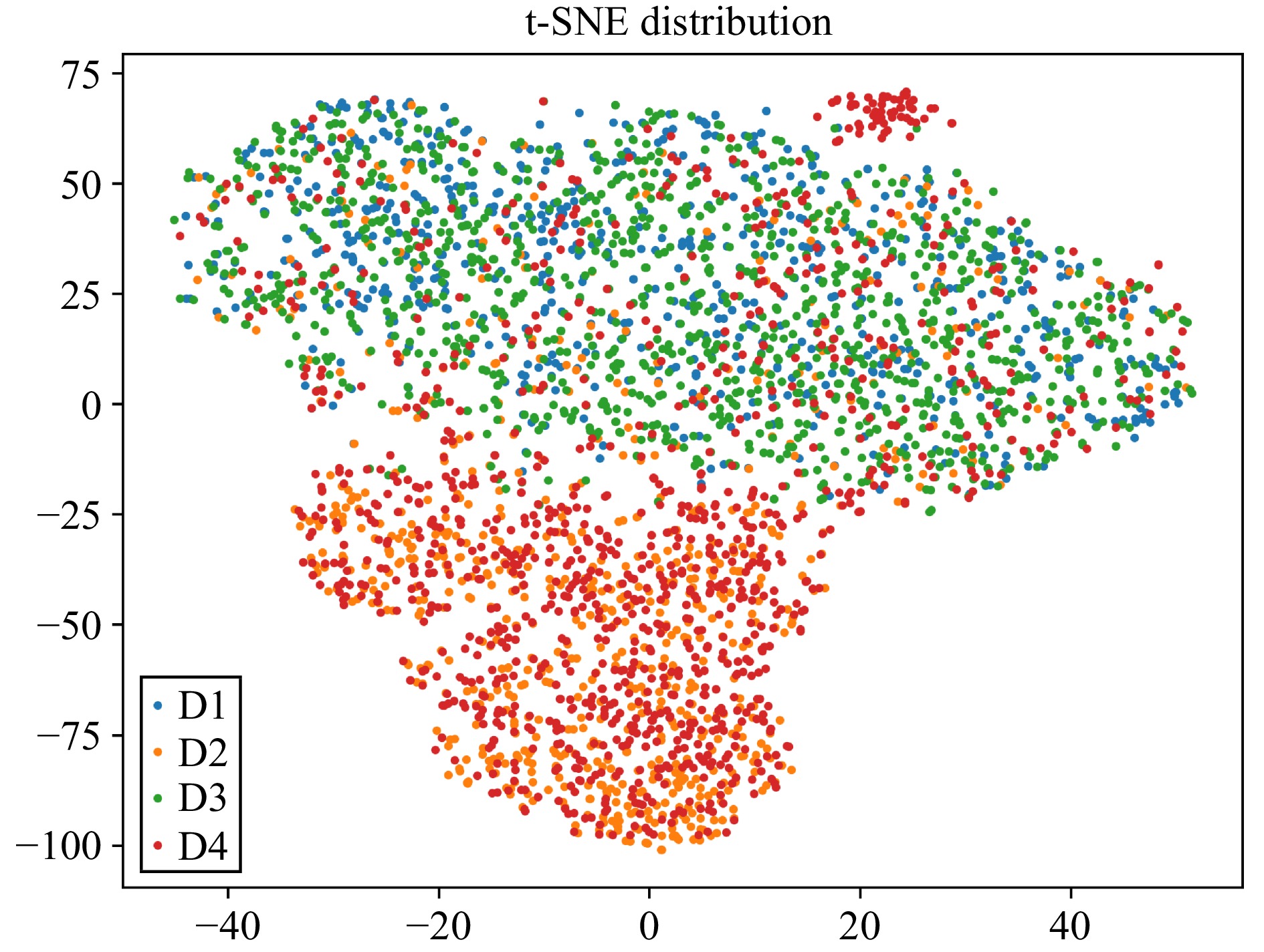
L. Van der Maaten and G. Hinton, “Visualizing data using t-SNE.,” J. Mach. Learn. Res., vol. 9, no. 11, pp. 2579–2605, 2008.
|
| [53] |
B. Fuglede and F. Topsoe, “Jensen-shannon divergence and hilbert space embedding,” in Proc. Int. Symp. Infor. Theory, ISIT 2., 2004, pp. 145–151.
|
| [54] |
Z. Liao, S. Wan, Y. He, and Q. Zou, “Classification of small GTPASES with hybrid protein features and advanced machine learning techniques,” Curr. Bioinf., vol. 13, no. 5, pp. 492–500, 2018. doi: 10.2174/1574893612666171121162552
|
| [55] |
L. Breiman, “Random forests,” Mach. Learn, vol. 45, no. 1, pp. 5–32, 2001. doi: 10.1023/A:1010933404324
|
| [56] |
M. A. Hearst, S. T. Dumais, E. Osuna, J. Platt, and B. Scholkopf, “Support vector machines,” IEEE Intell. Syst. Appl., vol. 13, no. 4, pp. 18–28, 1998. doi: 10.1109/5254.708428
|
| [57] |
T. Chen, T. He, M. Benesty, et al., “XGboost: Extreme gradient boosting,” R Package Version 0.4-2, vol. 1, no. 4, pp. 1–4, Jan, 2015.
|
| [58] |
L. Hu, P. Hu, X. Luo, X. Yuan, and Z.-H. You, “Incorporating the coevolving information of substrates in predicting HIV-1 protease cleavage sites,” IEEE/ACM Trans. Comput. Biol. Bioinform., vol. 17, no. 6, pp. 2017–2028, 2020. doi: 10.1109/TCBB.2019.2914208
|
| [59] |
X. Wang, W. Yang, Y. Yang, Y. He, J. Zhang, L. Wang, and L. Hu, “PPISB: A novel network-based algorithm of predicting protein-protein interactions with mixed membership stochastic blockmodel,” IEEE/ACM Trans. Comput. Biol. Bioinform., vol. 20, no. 2, pp. 1606–1612, 2022.
|
| [60] |
L. Hu and K. C. Chan, “Extracting coevolutionary features from protein sequences for predicting protein-protein interactions,” IEEE/ACM Trans. Comput. Biol. Bioinform., vol. 14, no. 1, pp. 155–166, 2017. doi: 10.1109/TCBB.2016.2520923
|
| [61] |
B.-W. Zhao, L. Wang, P.-W. Hu, L. Wong, X.-R. Su, B.-Q. Wang, Z.-H. You, and L. Hu, “Fusing higher and lower-order biological information for drug repositioning via graph representation learning,” IEEE Trans. Emerg. Topics Comput., vol. 12, no. 1, pp. 163–176, 2023.
|
| [62] |
X. Su, L. Hu, Z. You, P. Hu, L. Wang, and B. Zhao, “A deep learning method for repurposing antiviral drugs against new viruses via multi-view nonnegative matrix factorization and its application to sars-cov-2,” Brief. Bioinform., vol. 23, no. 1, p. bbab526, 2022. doi: 10.1093/bib/bbab526
|
Figures(6) / Tables(6)






 DownLoad:
DownLoad: